|
Chemical space is vast. So vast, that screening even a relatively small portion of this space within a drug discovery campaign is close to impossible. Just considering molecules with 30 heavy atoms consisting of carbon, oxygen, nitrogen, and sulfur we would find more than 1060 possible combinations [1] - a number that make even industry-scale million-member libraries of compounds appear small. While these libraries are still successfully used in high-throughput screens, a different approach, coined fragment-based drug discovery (FBDD) over 20 years ago [2], makes use of collections of so-called fragments with lower molecular weight (<300 Da) and a limited number of structural features [3]. The small size of the fragments drastically decreases the number of possible molecules and enables FBDD approaches to cover broader chemical space more efficiently. Due to their low complexity and low number of ‘non-essential’ atoms, screening fragments usually yields higher hit rates of molecules whose interactions with the target are defined by only one or two high quality contacts and high ligand efficiency (binding free energy per heavy atom). Accordingly, FBDD routines can be instrumental in finding druggable ‘hot spots’ and defining starting points for lead discovery. Yet, with small size often comes low affinity. Probing weak protein-fragment interactions demands for highly sensitive biophysical techniques, such as NMR, X-ray crystallography, surface plasmon resonance (SPR) or thermal shift assay (TSA), which often depend on availability of the purified target and yield only limited information on the effect the targeted site has on the functionality of the protein in a physiological environment [3]. The latter is particularly important for allosteric proteins considering the diverse mode of actions allosteric modulators can exert. Functional cell-based assays would allow for such a read-out in the absence of soluble target protein but are limited by the high concentrations of the fragments necessary to induce a measurable effect. One way to mitigate the challenges of detecting low affinity fragments, is so-called covalent tethering. In equipping fragments with a warhead that can form a covalent bond close to the target’s binding site, tethering increases the effective local concentration and therefore also apparent affinity. Reversible disulfide-bond formation between a thiol-modified fragment and a free naturally occurring or genetically inserted cysteine on the target is probably the most frequently used method to trap interacting fragments at the target site [4]. Most prominent example for this approach is the discovery of a covalent allosteric inhibitor for K-Ras G12C [5]. Recently, this approach has also enabled validation of the mode of action of a low-affinity allosteric fragment for PTP1B [6]. However, cysteines often represent critical sites for disulfide bond formation, posttranslational modifications and can be highly abundant throughout the target and off-target structures. Accordingly, disulfide-based tethering can often not be applied in cell-based assays and requires careful optimization and validation. In particular, if the cysteine is introduced by mutagenesis. In a recent paper, Mattheisen et al. [7] present an alternative elegant approach that leverages bioorthorgonal coupling chemistry to tether ligands to the class A GPCR C-C chemokine receptor 5 (CCR5) for functional fragment screening in live cells. Due to its critical role in HIV-1 pathogenesis, CCR5 has been rigorously characterized in terms of structure, function and druggability. Likewise, allosteric antagonists and the corresponding allosteric binding site have already been identified making it an ideal system to establish the proposed method [8]. While no large-scale fragment screening is conducted, the study provides a proof-of-concept and explores important considerations when establishing such a system. 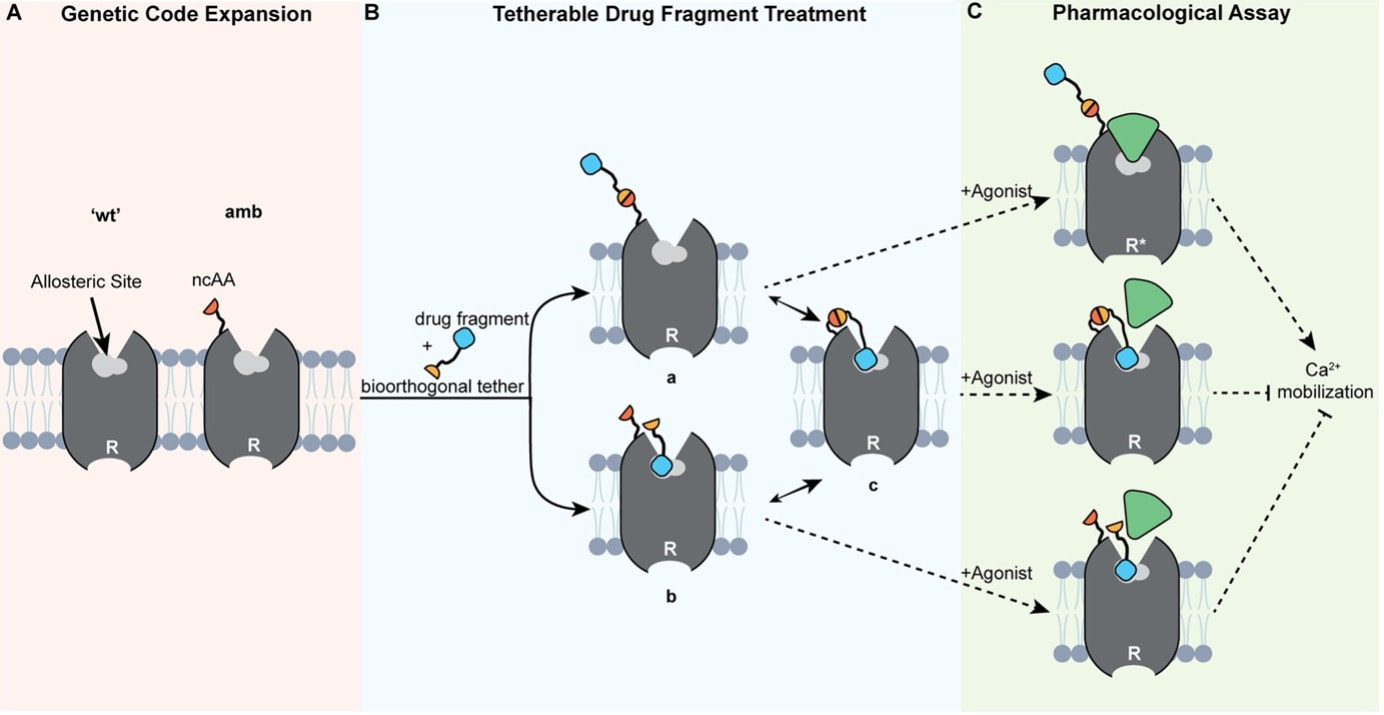 Figure 1: Setup to test antagonist activity of heterobifunctional ligands upon bioorthorgonal covalent tethering to a reactive handle incorporated into the GPCR CCR5. A) The reactive handle (here a non-canonical amino acid, ncAA) is incorporated into the target receptor close to the allosteric pocket by genetic code expansion. B) The heterobifunctional ligand is added and either a) first reacts with the handle, followed by binding or b) first binds to the pocket and then reacts or c) both. C) Potency is the assessed by adding an agonist and measuring activity of the receptor (here by Ca2+ mobilization). Figure and caption were adapted from Mattheisen et al. [7]. The system consists of a chemical handle that is incorporated into the receptor close to the allosteric binding site (Figure 1A) that can react with a reactive functional group under bioorthorgonal conditions compatible with mammalian cell culture (as introduced by the Bertozzi lab [9]). The reactive warhead is linked to a ligand via a PEG spacer, allowing a potential, now covalently coupled, antagonist to reach into the allosteric pocket which lays deeply buried in between the receptor’s transmembrane helices (Figure 1B). Functional response upon reaction with this heterobifunctional ligand is then probed by assessing cellular response to an agonist (Figure 1C). Using a combination of molecular dynamics simulations and subsequent experimental validation, Mattheisen et al. optimize assay conditions and controls by varying insertion points of the chemical handle in the receptor, warhead chemistry and linker length. Throughout the optimization process, the FDA-approved CCR5 antagonist maraviroc (mvc) is used as an active antagonist on the heterobifunctional ligand, which worked nicely for showing the theoretical potency increase the covalent tethering yields. So far so good. But mvc is a highly potent antagonist with nanomolar activity after all. So how would this system perform for lower affinity compounds, such as fragments? To test the capability of the system to distinguish between non-binders and low-affinity binders, six heterobifunctional ligands carrying low affinity mvc analogues were synthesized and tested in two cell-based assays, one testing for ability to block agonist activity and the other for blocking labeling of the reactive handle by a warhead-fluorophore conjugate. Intriguingly, this strategy revealed that only those heterobifunctional ligands whose low affinity mvc analogues show antagonist activity would also block labeling of the reactive handle by the fluorophore. Moreover, only the receptor containing the reactive label was inhibited. This is an important observation, since it indicated that on the one hand, the potency of low affinity compounds is effectively increased by covalent tethering and that on the other hand, affinity of the fragment for the pocket is the driving force for the reaction. In summary, the paper presents an innovative approach for live-cell functional screening of low-affinity compounds that adds another valuable method to the drug discovery toolbox for pharmaceutically interesting GPCR targets. However, some open questions remain: How easily is this system transferable to other receptor classes? How does the system perform in large-scale fragment screenings as shown for disulfide-tethering? How does it perform in a direct comparison with biophysical fragment screening in terms of hit-rates and false-positive rates? Can it facilitate fragment-to-lead discovery? References
1. Bohacek, R. S., McMartin, C. & Guida, W. C. The art and practice of structure‐based drug design: A molecular modeling perspective. Med. Res. Rev. 16, 3–50 (1996). 2. Shuker, S. B., Hajduk, P. J., Meadows, R. P. & Fesik, S. W. Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 274, 1531–1534 (1996). 3. Erlanson, D. A., Fesik, S. W., Hubbard, R. E., Jahnke, W. & Jhoti, H. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov 15, 605–619 (2016). 4. Erlanson, D. A., Wells, J. A. & Braisted, A. C. TETHERING: Fragment-Based Drug Discovery. Annu Rev Bioph Biom 33, 199–223 (2004). 5. Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013). 6. Keedy, D. A. et al. An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering. Elife 7, e36307 (2018). 7. Mattheisen, J. M. et al. Bioorthogonal Tethering Enhances Drug Fragment Affinity for G Protein-Coupled Receptors in Live Cells. J Am Chem Soc (2023) doi:10.1021/jacs.3c00972. 8. Tan, Q. et al. Structure of the CCR5 Chemokine Receptor–HIV Entry Inhibitor Maraviroc Complex. Science 341, 1387–1390 (2013). 9. Prescher, J. A., Dube, D. H. & Bertozzi, C. R. Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877 (2004).
1 Comment
|
- Home
- People
- Research
-
Training
-
Training Events
>
- 1st Workshop and PhD Induction Course
- 1st Training School & Networking Meeting
- Allostery in Drug Discovery Awareness Event and Symposium
- 2nd Training School & Networking Meeting
- IPR Training for Researchers & ESR Presentations on the Progress of their Research
- 3rd Training School & Networking Meeting
- European Conference in Allostery in Drug Discovery
- Secondments
- ALLODD Webinars
- Courses & Lectures
- Journal Club
-
Training Events
>
- Dissemination
- Events
- Newsroom
- Job Openings
- Blog
- Contact
 RSS Feed
RSS Feed
